
2024 Revisions to the Department of Health and Human Services (HHS)
Mandatory Guidelines for Urine Drug Testing
WFQA, LLC Regulatory Compliance Officer
INTRODUCTION
On October 12, 2023, the Department of Health and Human Services (HHS) revised the Mandatory Guidelines for Federal Workplace Drug Testing Programs using Urine (UrMG). The revisions are effective February 1, 2024. The key changes to the UrMG include:
- Establishing a process for HHS to annually publish the authorized drug testing panel (i.e., drugs, analytes, or cutoffs) and a biomarker testing panel for Federal workplace drug testing programs. In anticipation of this change, HHS on October 17th published a request for comments on adding fentanyl and its metabolite norfentanyl to the federal drug testing panel. And then, on Nov 17th, HHS revised its request for comments on the panel changes to include a proposal to remove MDMA and MDA from the federal drug testing panel. Comments on adding fentanyl and dropping MDMA/MDA for the federal drug testing panel were requested before the Dec 5th Drug Testing Advisory Board (DTAB) meeting. It will be DTAB that ultimately decides on the panel changes. The application of HHS MG drug testing panels to Department of Transportation (DOT)-mandated testing is not automatic and DOT will follow any DTAB decision with its own rulemaking process.
- Revising the definition of a substituted specimen to include specimens with a biomarker concentration inconsistent with that established for a human specimen. Currently a substituted specimen is defined as a urine specimen with creatinine and specific gravity values that are outside the physiologically producible ranges of human urine (creatinine <2mg/dL and specific gravity ≤1.0010 or ≥1.0200). In anticipation of adopting a biomarker testing panel for determining the validity of urine specimens, HHS has amended the definition of a substituted specimen as follows: Substituted Specimen. A specimen that has been submitted in place of the donor's specimen, as evidenced by the absence of a biomarker or a biomarker concentration inconsistent with that established for a human specimen, as indicated in the biomarker testing panel, or creatinine and specific gravity values that are outside the physiologically producible ranges of human urine, in accordance with the criteria to report a specimen as substituted.
- Raising the confirmatory test cutoff for morphine to 4000 ng/mL. Federal panel confirmation cut-offs for codeine and morphine have been 2000 ng/mL for over 20 years. The decision to raise the morphine cut-off to 4000 ng/mL was driven by what has been called "the poppy seed defense". For a positive codeine or morphine urine test, the MRO was required to consider poppy seed ingestion as a reasonable and acceptable explanation, unless the quantitation of codeine or morphine was ≥15,000 ng/mL, or there was "clinical evidence" of unauthorized, illicit use of codeine or morphine. This clinical evidence was obtained either from a physical examination of the donor (e.g. track marks, injection sites, etc.) or the donor's admission that he had used codeine or morphine without a valid prescription or medical administration of the drug. HHS has presented data that supports a urine specimen not having greater than 2000 ng of codeine or 4000 ng of morphine from ingestion of foods containing poppy seeds.
- Revising the Medical Review Officer (MRO) verification process for positive codeine and morphine urine specimens. In concert with raising the morphine cut-off to 4000 ng/mL as discussed above, HHS removed the additional decision point for codeine and morphine positives and the additional requirement for clinical evidence of illegal opioid use. Thus, the MRO will report a confirmed positive codeine or morphine result as a verified positive unless the donor provides a valid prescription for medication or documentation that morphine or codeine were medically administered, as an explanation for the morphine or codeine positive test.
- Requiring MROs to submit semiannual reports to HHS on Federal agency specimens that were reported as positive for a drug or drug metabolite by a laboratory and verified as negative by the MRO. MROs who review and report test results conducted on federal employees in testing designated positions subject to the HHS UrMG must submit the reports to HHS. This does not apply to DOT or Nuclear Regulatory Commission (NRC) mandated tests. The statistical report is of specimens that were reported as positive for a drug or drug metabolite by a laboratory and verified as negative by the MRO. The report must not include any personally identifiable information for the donor and must be submitted by mail, fax, or other secure electronic transmission method within 14 working days after the end of the semiannual period (i.e., in January and July)
These revisions are to the HHS UrMG and have not been adopted by DOT as revisions to 49 CFR Part 40. It is not clear whether DOT will engage in a rulemaking process to adopt these changes for DOT-mandated testing. However, MROs should strongly consider following the revised HHS UrMG, effective Feb 1, 2024 for the non-federal urine drug tests they review, especially where state laws reference the HHS MG as the standard for workplace drug testing.
Fentanyl and Fentanyl Analogs-Prevalence and Challenges
Per the Centers for Disease Control, overdose deaths remain the leading cause of injury-related deaths in the United States1. Fentanyl and fentanyl analogs, play a major role in the opioid epidemic. Fentanyl, a man-made or synthetic drug with properties like opiate drugs such as morphine and heroin, is primarily used medically to manage severe pain, especially in cases where other opioids may not be effective. While opioid potency varies greatly, fentanyl is estimated to be 50 to 100 times more potent than morphine and significantly more potent than heroin (figure 1). In 2021, there were over 100,000 drug overdose deaths with 75% related to opioids. Of these, 82% of these deaths were due to the use of synthetic opioids, such as fentanyl and fentanyl analogs in addition to other similar compounds2.
A fentanyl analog is a substance that is structurally similar to fentanyl but may have variations in chemical structure. These compounds have similar pharmacological effects, including pain relief and analgesia. Fentanyl analogs have different potency levels, durations of action, and side effect profiles compared to fentanyl. Some analogs, such as carfentanil, can have a potency of up to 10,000 times more potent than morphine and 100 times more potent than the parent compound fentanyl, increasing the risk of overdose4. Some fentanyl analogs have existed for decades and have or had medical uses, whereas non-pharmaceutical analogs are increasing in prevalence and use. These compounds include sufentanil, alfentanil, remifentanil, carfentanil, α-methylfentanyl, 3-methylfentanyl, acetylfentanyl, furanyl fentanyl, butyrylfentanyl, acrylfentanyl, furanylfentanyl and β-hydroxythiofentanyl, and more. Further, N-phenyl-1-(2-phenylethyl)piperidin-4-amine (4-ANPP) has been designated by the Drug Enforcement Administration (DEA) as an intermediate chemical precursor used in the synthesis process for the illicit production of the schedule II controlled substance, fentanyl, and other related opioids.3
Figure 1. Relative Opioid Potency - Click Image to Enlarge
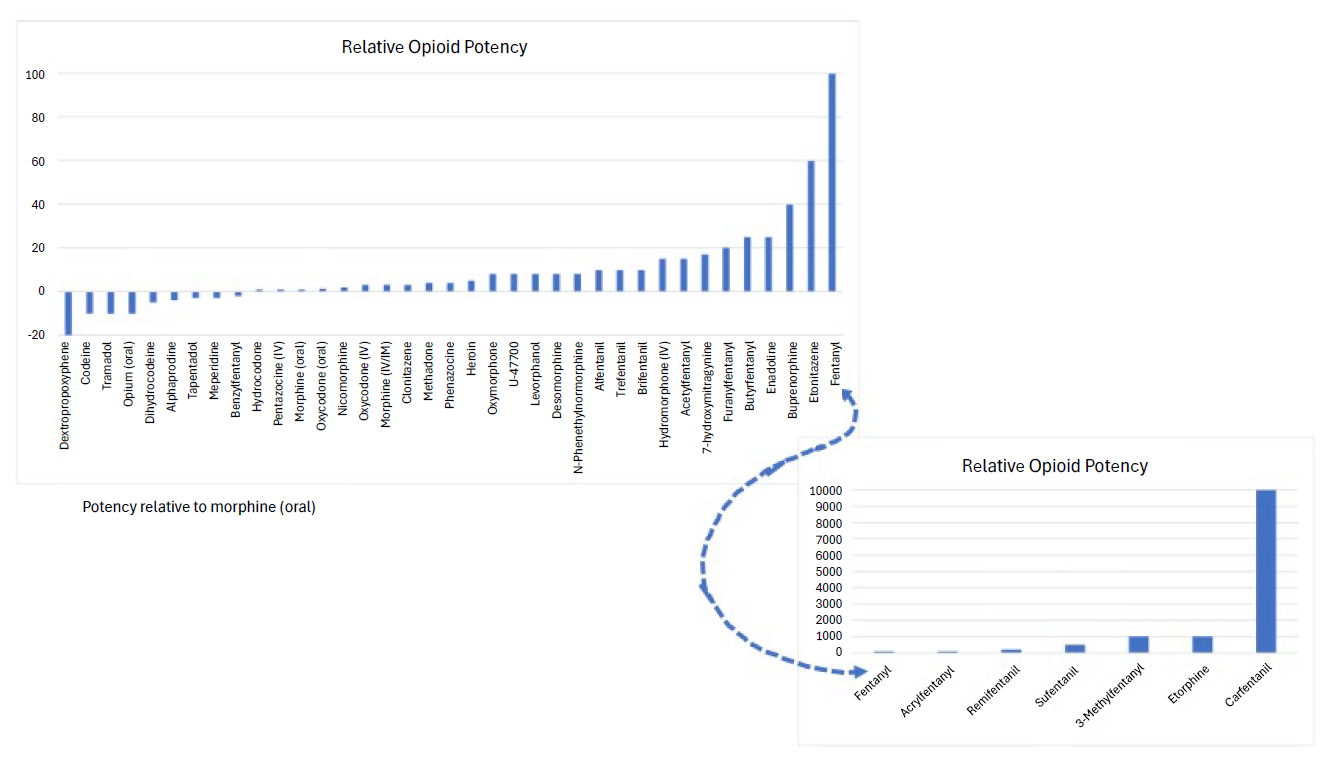
Illicitly produced fentanyl analogs have been a major concern in the context of the opioid epidemic, as they contribute to the increased risk of overdose and death. Authorities work to monitor and regulate these substances to address the public health risks associated with their use. The diversity in fentanyl analogs poses a challenge for forensic and clinical testing, workplace drug testing, public health officials, law enforcement and consumers as new variations emerge coupled with their varying legal statuses. An additional complicating factor is the combination of fentanyl with other drugs such as xylazine, methamphetamine, cocaine, methylenedioxymethamphetamine (MDMA), and other opioids. In many cases users may be unaware that the drugs they are consuming contain these extremely potent combinations and may include counterfeit pills mimicking legitimate therapeutic drugs5.
Testing of biological matrices such as blood, urine, oral fluid, etc. that may contain fentanyl and/or fentanyl analogs can be challenging. Depending on the scope of testing of a laboratory, these analytes may or may not be detected. Further, if a laboratory is utilizing an immunoassay test for presumptive or screening purposes, unless the test kit is specific for fentanyl, it will not detect these compounds. Therefore, laboratories would need a more specific test such as the use of mass spectrometry coupled with chromatography (i.e., GC-MS, LC-MS, LC-MS/MS, LC-TOF, etc.). As new, and emerging fentanyl analogs become available to consumers, testing laboratories must rely on analytical techniques such as high-resolution mass spectrometry to elucidate these compounds and expand their testing capabilities. Even in cases of fentanyl and fentanyl analog overdose, the concentration of these compounds can be extremely low and may be lower than the analytical capabilities of the testing laboratory.
As the opioid epidemic continues, employers and workplace drug testing laboratories must remain vigilant in the extreme impairment risks associated with the use of fentanyl, fentanyl analogs and the mix of these with other drugs.
REFERENCES
- Understanding Drug Overdoses and Deaths. Centers for Disease Control. https://www.cdc.gov/drugoverdose/epidemic/index.html (Accessed November 30, 2023)
- Ahmad FB, Sutton P. Provisional drug overdose death counts. 2021. https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm (Accessed November 30, 2023)
- Schueler HE. Emerging Synthetic Fentanyl Analogs. Acad Forensic Pathol. 2017 Mar;7(1):36-40. doi: 10.23907/2017.004. Epub 2017 Mar 1. PMID: 31239954; PMCID: PMC6474477.
- Jalal H, Burke DS. Carfentanil and the rise and fall of overdose deaths in the United States. Addiction. 2021 Jun;116(6):1593-1599. doi: 10.1111/add.15260. Epub 2020 Sep 25. PMID: 32935381; PMCID: PMC8019064
- Drug Fact Sheet, Counterfeit Pills. Drug Enforcement Agency. https://www.dea.gov/sites/default/files/2021-05/Counterfeit%20Pills%20fact%20SHEET-5-13-21-FINAL.pdf (Accessed December 7, 2023)
Insights into the DOT Oral Fluid Rule for 49 CFR Part 40 and Its Impacts on Medical Review Officers for Oral Fluid Testing
Senior Policy Executive Advisor, National Drug & Alcohol Screening Association
Mandatory Disclaimer: I am an employee of the United States Department of Transportation (DOT) on detail to the National Drug and Alcohol Screening Association (NDASA) as the Senior Policy Executive Advisor. The views reflected here are expressed in my personal capacity. My views have not been subjected to review, clearance, or approval by DOT, and they do not necessarily represent the views of DOT.
In the last issue of this newsletter, we covered many of the elements of the DOT's Oral Fluid Final Rule, 88 Federal Register 27596 (May 2, 2023), which became effective on June 1, 2023. In that article, we emphasized the aspects of the final rule that were not related to actual DOT-regulated oral fluid testing. In this article, we will dig deeper into some aspects of the final rule to implement DOT-regulated oral fluid testing throughout 49 CFR Part 40 (Part 40) and how they will impact MROs for oral fluid testing and the DOT's reasoning. As we get closer to the HHS-certification of laboratories to perform oral fluid testing and the identification of what devices those laboratories will use, the actual provisions of the final rule for implementing oral fluid testing will become more relevant.
In the DOT oral fluid rulemaking, there were 417 commenters who filed literally thousands of individual subject matter comments. The commenters, including MROCC, and individual MROs, submitted excellent comments that provided insight, guidance, affirmation, and sometimes caused changes to be made. Here are some of the relevant provisions:
MRO Training for Oral Fluid testing
The DOT decided not to require additional training for MROs to review oral fluid test results. The addition of oral fluid testing will not change the way MROs perform their responsibilities under Part 40. The MRO will continue conferring with laboratories, verifying test results by interviewing donors, and the other basic aspects of MRO practice will remain unchanged.
DOT acknowledged comments that said MRO training already covers the science of oral fluid drug testing. Consequently, DOT suggested that "MRO training organizations offer oral fluid modules to augment the training of MROs who are already current on their training certification requirements...[and] strongly suggest MROs seek supplemental information about oral fluid testing by the time HHS certifies at least two laboratories to conduct oral fluid testing." 88 Federal Register 27619-20 Thus, MROs are not required to obtain additional training before their next certification date.
Stand Down
Under Part 40 and the corresponding DOT agency regulations, an employer can submit a waiver request to "stand down" an employee from performing safety-sensitive functions based on a laboratory confirmed non-negative result (i.e., positive, adulterated, substituted or any combination thereof) before and until the MRO issues the employer a verified result. The process to receive a stand down waiver is extensive and the authority granted to the employer is limited. The existence of stand down waivers under 49 CFR § 40.21 has become extraordinarily rare, with only 2 or 3 stand down waivers existing today.
If a stand down waiver is in place, the MRO could issue a verified negative result due to a legitimate medical explanation. If the MRO issues a verified negative result, the DOT wanted to ensure the employer would not send the employee back in for another test, using another allowable testing methodology. To prevent this, DOT added 49 CFR § 40.21(c)(2)(vii)(C). DOT's rational was "We did not want the employer to order a second test using a different methodology to see if the window of detection could later impact the result." 88 Federal Register 27608
Laboratories will report oral fluid device expiration date to MRO
The laboratory must add oral fluid collection device expiration date to the results report it generates for the MRO under 49 CFR § 40.97(c)(1)(i)(I). This data is important to the MRO for seeing whether the device was expired at the time of the collection, which would be a fatal flaw. If a laboratory provides the MRO with Copy 1 of the CCF, the expiration date will already be recorded there. This change applies only where a laboratory wants to report negative results to an MRO in report format instead of using Copy 1. 88 Federal Register 27619
MRO Review of negative results for urine and oral fluid
In practices where MRO staff review and affix the MRO's signature to the negative results, MROs will only need to review 5% or a total of no more than 500 negative results "of all specimen types combined" in any quarter under 49 CFR § 40.127. DOT chose not to raise the number to 500 reviews for urine plus 500 reviews for oral fluid because there was no reason to impose this cost increase for the MRO review process. 88 Federal Register 27620
Reporting results from a urine and then an oral fluid test
It is anticipated to be infrequent for an employee to provide both a urine specimen and an oral fluid specimen that both go to the laboratory, but it will happen. When an employee provides a suspect urine specimen (e.g., temperature out of range, excess foaming, etc.), and then undergoes a successful oral fluid specimen collection, or vice versa, the collector would send both specimens to the respective laboratories for testing. In this scenario, as when the MRO receives two separate urine specimen results for the same testing event now, the MRO will continue to report the multiple verified results from one testing event in accordance with 49 CFR §40.162. Following through with this section, if there are two negative results, the MRO would report a single negative result to the employer for the testing event. If there is a negative and a verified non-negative result, the MRO would report only the verified non-negative result to the employer. For more discussion, see 88 Federal Register 27625.
Dry Mouth Evaluations
The decision of whether to administer a urine or an oral fluid test is the decision of the employer. The DOT encourages the employer to develop a standing order to provide instruction for the collector to know whether to administer a urine or an oral fluid test by type of test and in problem collections, such as an insufficient specimen. (Here is an example of a standing order.) An employer has the option to direct its collector to change from one methodology to another when the employee provides an insufficient specimen. Hopefully, this will greatly decrease the number of shy bladder evaluations that need to be performed.
When a donor is unable to provide a sufficient amount of oral fluid, the situation is referred to as a "dry mouth" scenario and it is addressed in 49 CFR § 40.193, like its cousin the "shy bladder". As in a shy bladder scenario, if the donor fails to provide a sufficient specimen by the end of the allotted time (one hour for oral fluid testing), the DER must consult with the MRO before sending the donor to obtain, within 5 days, an evaluation from a licensed physician, acceptable to the MRO, who has expertise in the medical issues raised by the employee's failure to provide a sufficient specimen. For this requirement and the other aspects of dry mouth scenarios, the DOT attempted to create parallel provisions to the shy bladder procedures in section 40.193.
In §40.193(e), DOT offers some examples of conditions that might be medical explanations of providing an insufficient quantity of oral fluid. It also provides examples that would not constitute a valid medical explanation such as unsupported assertions of "situational anxiety" or dehydration. DOT was confident that, as fully qualified physicians, MROs can "assess the legitimacy of the conditions underlying an individual's inability to provide a sufficient specimen under any approved testing methodology."88 Federal Register 27624
If a collection begins with one specimen methodology but results in an insufficient specimen collection, and the employer allows a second methodology to be used that results in a sufficient specimen collection, there will be a test result and no need to go through a shy bladder or dry mouth evaluation. If the second methodology does not yield a sufficient specimen, then the donor will have an evaluation for ONLY THE SECOND insufficient specimen type attempted. DOT explained: "To be clear, the employer must send the employee for only a dry mouth medical evaluation if the employee has not provided a sufficient oral fluid specimen following an insufficient urine specimen. The MRO will only proceed with the dry mouth evaluation and not proceed with the shy bladder evaluation. Similarly, the employer must not send the employee for a dry mouth evaluation if the employee has not provided a sufficient urine specimen following an insufficient oral fluid specimen. The MRO will only proceed with the shy bladder evaluation and not proceed with the dry mouth evaluation. Only a shy bladder medical evaluation is to be done at that point." 88 Federal Register 27625
Remember, oral fluid testing for DOT will only be allowed when the Department of HHS certifies two laboratories for oral fluid testing - a primary laboratory and a secondary laboratory.
Cannabis Descheduling and the Demise of Section 280E
Although the final decision rests with the DEA, if cannabis is reclassified to Schedule III, marijuana growers, processors, transporters or sellers (marijuana-related businesses or MRBs) will cease being encumbered by onerous operating and taxing obstacles to become significantly more profitable, federally funded cannabis research will finally occur, and investment in marijuana-related businesses should soar.
Causing a day-long, double-digit cannabis stock surge peaking at 40.56%, on Aug. 30, the U.S. Department of Health and Human Services (HHS) recommended that the U.S. Drug Enforcement Administration (DEA) reschedule marijuana from Schedule I to Schedule III of the Comprehensive Drug Abuse Prevention and Control Act of 1970, 21 U.S.C. Sections 801, Et. Seq (1970) (Controlled Substance Act).
Impact of Cannabis Murky Legality and Scheduling
While legal in 38 states and projected to generate $33.5 billion in 2023 legal sales, the Controlled Substance Act currently lists marijuana next to heroin as a Schedule I controlled substance having "a high potential for abuse" and for which there's "no currently accepted medical use in treatment" and "a lack of accepted safety for use" "under medical supervision." See 21 U.S.C. Section 812(b)(1).
Tetrahydrocannabinol is marijuana's psychotropic-effect producing component and, whether deemed "medical" (purchasable only with state-issued card to treat resident's statutorily defined "covered medical condition") or adult-use (purchasable by anyone over 21 from any state with a valid identification), cannabis takes four forms: flower that is smoked; oils ingested by vaporizing; concentrates only consumable after being heated to a high temperature; and infused products ranging from eye drops to edibles. With few exceptions, medical and adult-use cannabis items are identical and only delineated by their purchasers: medical card patients; or adult use consumers.
The Controlled Substance Act prohibits marijuana's cultivation, distribution, dispensation and possession and, pursuant to the U.S. Constitution's supremacy clause, state laws conflicting with federal law are generally preempted and void. U.S. Const., Art. VI, cl. 2; Wickard v. Filburn, 317 U.S. 111, 124 (1942)("No form of state activity can constitutionally thwart the regulatory power granted by the commerce clause to Congress").
Pursuant to The Controlled Substance Act, the DEA "schedules" drugs from most perilous - Schedule I - to least harmful - Schedule V - based on three factors: potential for abuse: how likely is this drug to be abused; accepted medical use: is this drug used as a treatment in the United States; and safety and potential for addiction: Is this drug safe, how likely is it to cause addiction and, if so, what kinds of addiction?
Beyond making cannabis 100% federally illegal and preventing it from being sold outside of each respective legalized-marijuana state (i.e., no "interstate cannabis commerce"), Schedule I classification both precludes any federally funded cannabis research and imposes heinous tax consequences on marijuana-related businesses.
Under Section 280E of the Internal Revenue Code, state-legal MRBs are forbidden from taking any tax deduction or credit other than "cost of goods sold." See 26 U.S. Code Section 280E (1982). Specifically, Section 280E provides:
"No deduction or credit shall be allowed for any amount paid or incurred during the taxable year in carrying on any trade or business if such trade or business (or the activities which comprise such trade or business) consists of trafficking in controlled substances (within the meaning of schedule I and II of the Controlled Substances Act) which is prohibited by Federal law or the law of any State in which such trade or business is conducted."
Thus, unlike every other legitimate industry, MRBs are denied ordinary and necessary business expense tax deductions (e.g., wages, rent, utilities, and insurance) causing them to be vastly more expensive, and dramatically less profitable, to operate.
By way of background, domestic "contraband taxation" commenced with 1760's Navigation Acts banning Dutch tea, requiring colonists to purchase British tea through the East India Co. (thereby conferring a trade monopoly) and taxing Dutch tea merchants and possessors at an exorbitant rate. Similarly, in 1919, Congress ratified the Eighteenth Amendment outlawing alcoholic beverages transportation and sale and enacted the Volstead Act assessing a $1,000 fine/tax per every unlawful alcohol trafficking transaction. Further, in 1937 Congress adopted the Marihuana Tax Act requiring those producing, possessing, or selling cannabis to obtain a tax stamp demonstrating the payment of a cannabis tax.
Up until California and Arizona's medical use legalization in 1996, Section 280E had not been applied to state-legal cannabis causing a coalition of federal agencies to issue a federal directive that:
"To the extent that state laws result in efforts to conduct sales of controlled substances prohibited by federal law, the IRS will disallow expenditures in connection with such sales to the fullest extent permissible under existing federal tax law."
Pursuant to this directive, the Internal Revenue Service (IRS) has strictly applied Section 280E against state-legal MRBs and the courts have supported the IRS' "state-legal cannabis constitute federally unlawful drug trafficking" argument. See Patients Mutual Assistance Collective Corp. v. Commissioner, (9th Cir. No. 19-73078 2021); Standing Akimbo v. United States, 955 F.3d 1146 (10th Cir. 2020); and Feinberg v. Commissioner of the Internal Revenue Service, 916 F.3d 1330 (10th Cir. 2019).
To understand Section 280E's impact on disallowing MRBs' ordinary and necessary business expense deductions, below are contrasting tax calculations of a muffin shop and cannabis dispensary experiencing identical revenue:
| ALM's Muffins | ALM's Dispensary | |
| Gross Revenue: | $1,000,000 | $1,000,000 |
| Minus Costs of Goods Sold: | ($550,000) | ($550,000) |
| Minus Business Expenses: | ($350,000) | ($0.00 allowed) |
| Taxable Income: | $100,000 | $450,000 |
| Federal Tax: | $39,000 | $175,500 |
Although the muffin shop would realize $61,000 of net income, after the $350,000 of "nondeductible" wages, rent, utilities and insurance costs is factored in, the dispensary would incur a $75,500 net loss.
Thus, through denying MRBs' ordinary and necessary business expense tax deductions, Section 280E places MRBs at a ridiculous competitive disadvantage and virtually precluding them from achieving profitability.
Seismic Impact of Reclassifying Cannabis on Research, Operations and Investment
If the DEA reclassifies Marijuana from Schedule I to Schedule III, federally funded cannabis research will finally occur, MRBs will cease being encumbered by Section 280E, and investment in MRBs will skyrocket.
First, reclassifying to Schedule III indicates that the federal government deems cannabis to have "moderate to low potential for physical and psychological dependence" (and not Schedule I's "no current medical use with high potential for abuse or addiction" stigma) opening the floodgates for federally-funded research. Use-in-treatment data from 38 state's medical markets and a staggering body of international scientific literature already supports cannabis' medical efficacy and treatment for conditions ranging from cancer and post-traumatic stress disorder to opioid use mitigation. Federally approved and funded research will both enhance public acceptance of cannabis medical application and deploy the Feds nearly infinite resources like those of the U.S. Food and Drug Administration (FDA).
Budgeted at $6.1 billion, the FDA protects and promotes public health through supervising food safety, tobacco products, dietary supplements, prescription and over-the-counter pharmaceutical drugs, cosmetics, animal foods and feed, and veterinary products. Federal Food, Drug, and Cosmetic Act, 21 U.S.C. 301, et seq.. Reclassifying to Schedule III will empower the FDA to regulate those growing, processing, selling or transporting cannabis and promulgate uniform standards and prohibitions.
Second, reclassifying to Schedule III will remove Section 280E's prohibitions and allow marijuana-related businesses to take ordinary and necessary business expenses tax deductions resulting in cost reduction, ease of operations and an increase of profit.
Third, as a result of federally-funded research, wider public acceptance, and higher profitability, reclassifying to Schedule III will enhance cannabis investment. While not likely to mirror the "Aug. 30, 2023 cannabis stock surge," reclassification will enhance the valuation of an industry beset by a lack of investment capital, a mishmash of 38 conflicting regulatory bodies, and an inability to meet exceedingly optimistic performance goals.
Path to Cannabis Schedule III Classification
While reclassification to Schedule III will not remove cannabis' federal illegality, it is a significant step toward ending prohibition and bolstering a nascent industry.
The final determination rests with the DEA, which, based on an eight-factor analysis, will weigh whether the HHS scheduling recommendation, scientific and medical evaluation, and all other relevant data constitute substantial evidence that marijuana should be reclassified to Schedule III.
If determining that rescheduling is appropriate, the DEA will issue a proposed rule followed by a notice and comment period.
Although whether MRBs will receive a future "tax credit" for past Section 280E disallowed business expenses tops the list of rescheduling's unanswered questions, the future remains uncertain pending the DEA determination and rule promulgation.
Reprinted with permission Steven M Schain. Copyright ©2023 by Steven M. Schain, Esquire
Presenting IAMRO: The International Association of Medical Review Officers
As chairperson of the above-noted organization, I am pleased to introduce our recently formed not-for-profit organization. I suspect this is a new entity to most readers of this newsletter, and as such, my intent here is to provide you with an overview of who we are, how we came to be, and our focus and initiatives.
Who We Are
IAMRO is a professional membership association of physicians and medical practitioners who provide Medical Review Officer (MRO) services in their country of practice, health professional Subject Matter Experts (SMEs), and forensic toxicologists who work in the field of workplace drug and alcohol testing.
How We Came to Be
In September 2022, at the International Forum for Drug and Alcohol Testing (IFDAT) conference in Salzburg, Austria, an Expert Panel discussion centered around international MRO networking. Presenters Prof. Dr. Serap Annette Akgür (Turkey), Dr. Monica Eneholm (Sweden), Dr. Trevor Maze (UK), and Dr. Todd Simo (USA), led a discussion which focused on the need for cross-border professional support, how a network could be achieved, considerations for moving forward, current examples, and on the establishment of an IFDAT MRO Working Group.
At the end of the session, due to extraordinary international attendee interest, multiple hallway conversations ensued, and by the end of that day, stakeholders from every part of the globe met. In that meeting, an informal working group sprang up. That group included members of the panel and interested conference attendees making a commitment to meet to begin the process of creating an international organization.
Overarching Tenets
The world is becoming very small. Global organizations are looking far and wide for vendors who can fulfill their requirements. Our vision is to have a network of knowledgeable experts who provide consistency by "following the sun", allowing organizations to leverage the same drug-free workplace rules regardless of where they operate geographically. Having "In-Region" MRO services is logically efficient, linguistically optimal, and affords country-specific rules to be handled by local experts, with an international resource base, as need be. The overarching tenet is to create a standard to enable organizations who operate globally to have confidence in their application of an accredited/certified approach that is consistent with in-region medical expertise, as it relates to their individual drug-free workplace programs.
Our Focus and Initiatives
Since September 2022, a dedicated group of individuals from across the world have met regularly with the intent of creating a draft International Standard for MROs concerning required activities and responsibilities, and clarifying the numerous components of the verification process. Since only three countries have a codified MRO accreditation in place, the basis for the Standard were the Australia/New Zealand model and US DOT 49 CFR Part 40, Subpart G - Medical Review Officers and the Verification Process. The focus was to organize the information in a logical sequence, formatted for readability, and written in plain English to be easily interpreted by anyone, regardless of their language of origin. The elements for "standardization" include strong recommendations for two contributing components that are not in the scope of MRO practices: collection practices where we urge utilization of chain of custody procedures; and laboratory analyses where we encourage consistent and valid scientific methodologies. For the medical review procedures, we defined the standards to be utilized by MROs in the performance of their work. We specifically covered the topics of the review of the external chain of custody, interpretation of the laboratory result, donor contact, donor interview, verification of the results and reporting to the employer. The overarching goal is to offer employers drug testing programs that are fair and consistent with due processes for all parties.
Our Starting Point and Process
Our model prioritized collaboration of like-minded and motivated SMEs from around the world. As noted previously, our priority was to have a document that is written in plain English and is understandable. Our meetings were facilitated by George Gilpatrick and me, after collective agreement on the approach to be used and priorities of the group. Our ongoing discussion and debate included peer review, consensus on the final draft, and fine-tuning along the way so that after 28 iterations, we had a finalized document for presentation.
Administratively, the International Association of Medical Review Officers has been registered as a not-for-profit organization in the US and is recognized as doing business as IAMRO. The Board of Directors was formalized in August of 2023 and the first Board meeting was held prior to the European Workplace Drug Testing Society (EWDTS) conference.
Board Members
Prof. Dr. Serap Annette Akgür
Dr. Charles Appleton
Dr. Monica Eneholm
George Gilpatrick
Joe Lofgren
Dr. Trevor Maze
Jo McGuire
Stefan Nicolaidis
Dr. Wim Schielen
Dr. Todd Simo
Nadine Wentzell
Board Executive
MRO - Dr. Todd Simo
Treasurer - Joe Lofgren
Secretary - George Gilpatrick
Chairperson - Nadine Wentzell
Our Vision
"Creating a future where consistency, fairness, and scientific rigor of the medical review process in workplace drug testing becomes the global norm, transcending borders, boundaries, and cultures for safer, healthier, and more productive work environments worldwide."
Our Mission
"To establish the standard for workplace drug testing worldwide, by providing rigorous tools, comprehensive training, and advocating for fairness through scientific integrity. We are committed to facilitating international procedural consistency and empowering employers and Medical Review Officers to create healthier and safer work environments."
Proposed Standard - Key Areas for the MRO
Specific areas of the Standard include MRO qualifications, responsibilities, laboratory relationships, functions in review of laboratory results, donor notification, verification processes and prohibitions, "uncommon" situations, transmission of results, and administrative functions.
The draft Standard was finalized in June 2023. This document served as the basis for a presentation at EWDTS in Istanbul, Turkey, in October 2023, by Working Group members Dr. Trevor Maze, United Kingdom, Dr. Todd Simo, USA and me, Nadine Wentzell, Canada.
Next Steps
Several areas of accreditation or certification have been initiated and others are at various stages in the process. Presentation at EWDTS introduced the Standard with the goal of approval from that organization and subsequent acceptance within the European Union. Discussions regarding a submission to the European Accreditation Agency for application of ISO certification have begun, with additional areas yet to be explored.
The IAMRO has created the framework for international MRO training. The curriculum includes much of what has been identified as included in the Standard, with emphasis on what to do in situations of a positive test result review, "troubled test" review, and administrative functions of an MRO.
IAMRO as a Board is in the process of establishing our priorities for the coming year, creating initiatives to align with those priorities, and to grow our associate membership internationally.
Any MRO or associate/affiliate looking for more information on IAMRO and/or with interest in becoming an association member is invited to visit our IAMRO website at https://iamro.org.
 ODAPC - DOT
- Office of Drug & Alcohol Policy & Compliance (ODAPC)
- DOT 49 CFR Part 40 Procedures for Transportation Workplace Drug and Alcohol Testing Programs
- Subscribe to the ODAPC Updates and News
- 2024 DOT Random Testing Rates
SAMHSA - HHS
- Medical Review Officer Guidance Manual for Federal Workplace Drug Testing Programs
- Mandatory Guidelines for Federal Workplace Drug Testing Programs using Urine (UrMG)
- Mandatory Guidelines for Federal Workplace Drug Testing Programs using Oral Fluid (OFMG)
NRC
CUSTODY AND CONTROL FORMS (CCFs)
- 2020 Federal CCF for Urine and Oral Fluid Specimens
- 2020 Guidance for Using the Federal CCF for Urine Specimens



Medical Review Officer
Certification Council (MROCC)
3231 S Halsted St, Ste Front ###167
Chicago, IL 60608
Tel: 847.631.0599
Email: mrocc@mrocc.org
Editor: James Ferguson, DO
Managing Editor: Kristine Pasciak
©2023 Medical Review Officer Certification Council
ISSN: 2833-0870
MRO Quarterly is an educational publication intended to provide information and opinion to health professionals. The statements and opinions contained in this document are solely those of the individual authors/contributors and not MROCC. MROCC and its editorial staff disclaim responsibility for any injury to persons or property resulting from any ideas or products referred to in this newsletter.
To unsubscribe from MROCC emails, please send an email to mrocc@mrocc.org with the subject unsubscribe.